Commits on Source (32)
-
Nathalie Bechon authored82d771e9
-
aherrero authored44988b27
-
-
-
-
Jean CURY authored1204385b
-
-
-
-

-
-
-
-
-
-
-
 Remi PLANEL authored3f828210
Remi PLANEL authored3f828210 -
dcd4426c
-
-
Jean c authoredd14a9749
-
Jean c authoreda05c7aec
-
-
-
Jean CURY authoredc9e6d058
-
-
-
-
c00fff1a
-
-
-
-
Remi PLANEL authoredf5c030e4










Showing
- components/content/StructureDb.vue 4 additions, 3 deletionscomponents/content/StructureDb.vue
- content/2.general-concepts/5.defense-systems-discovery.md 10 additions, 1 deletioncontent/2.general-concepts/5.defense-systems-discovery.md
- content/2.general-concepts/7.mge-defense-systems.md 4 additions, 3 deletionscontent/2.general-concepts/7.mge-defense-systems.md
- content/3.defense-systems/abih.md 7 additions, 0 deletionscontent/3.defense-systems/abih.md
- content/3.defense-systems/card_nlr.md 11 additions, 11 deletionscontent/3.defense-systems/card_nlr.md
- content/3.defense-systems/drt.md 31 additions, 12 deletionscontent/3.defense-systems/drt.md
- content/3.defense-systems/fs_hp.md 11 additions, 9 deletionscontent/3.defense-systems/fs_hp.md
- content/3.defense-systems/gao_mza.md 11 additions, 9 deletionscontent/3.defense-systems/gao_mza.md
- content/3.defense-systems/gaps1.md 13 additions, 8 deletionscontent/3.defense-systems/gaps1.md
- content/3.defense-systems/lamassu-fam.md 16 additions, 26 deletionscontent/3.defense-systems/lamassu-fam.md
- content/3.defense-systems/lit.md 19 additions, 14 deletionscontent/3.defense-systems/lit.md
- content/3.defense-systems/mmb_gp29_gp30.md 10 additions, 10 deletionscontent/3.defense-systems/mmb_gp29_gp30.md
- content/3.defense-systems/mok_hok_sok.md 18 additions, 11 deletionscontent/3.defense-systems/mok_hok_sok.md
- content/3.defense-systems/shosta.md 14 additions, 10 deletionscontent/3.defense-systems/shosta.md
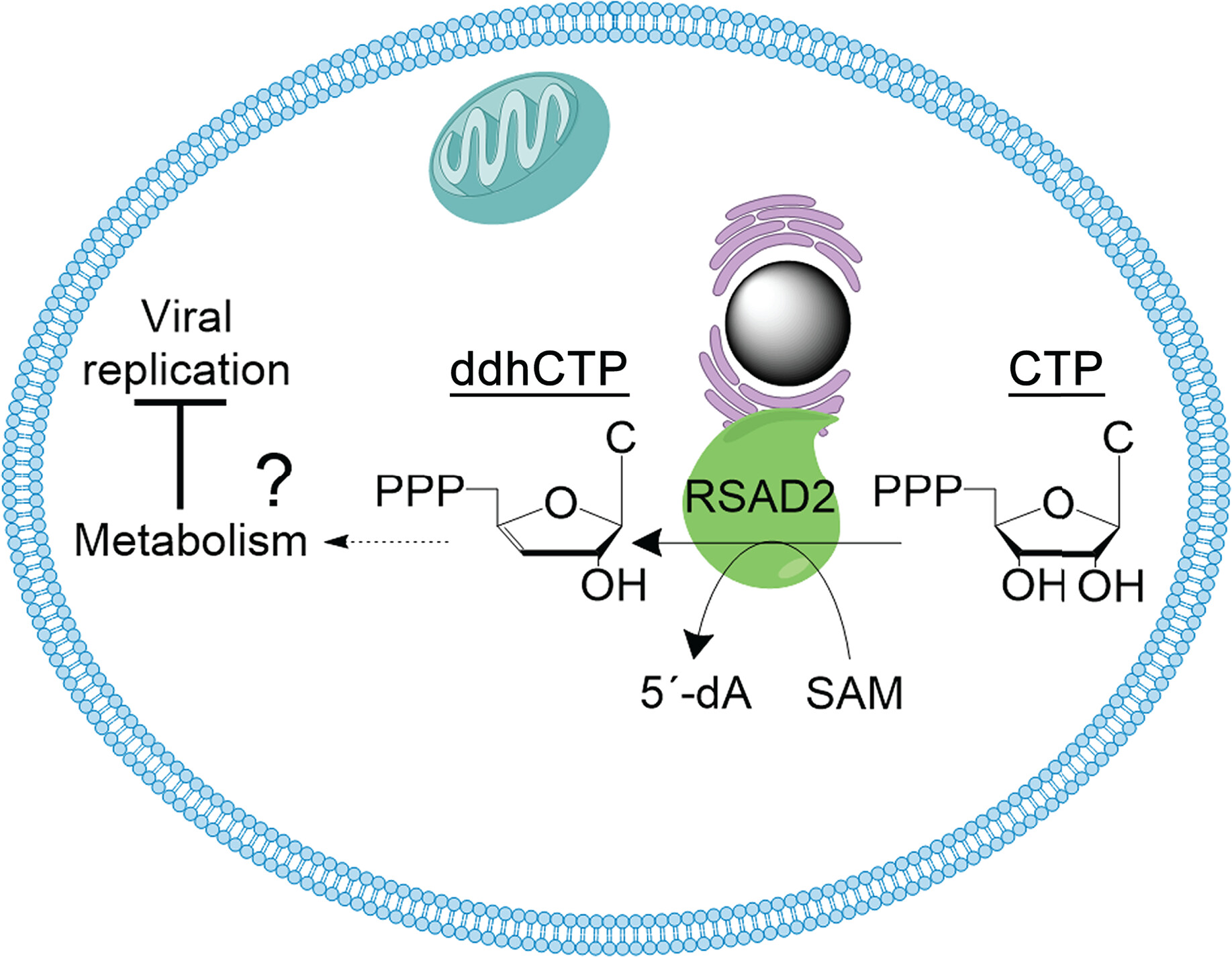
- content/3.defense-systems/viperin.md 12 additions, 17 deletionscontent/3.defense-systems/viperin.md
- public/viperin/human_vip.jpg 0 additions, 0 deletionspublic/viperin/human_vip.jpg
public/viperin/human_vip.jpg
0 → 100644
{kind=link}
340 KiB